What is Phylogeny?
|
Phylogeny is the study of evolutionary relationships between species, often organized into phylogenetic trees. Phylogenies are useful tools for understanding gene evolution, and can be constructed from species data such as morphological features, DNA, and protein sequences [1].
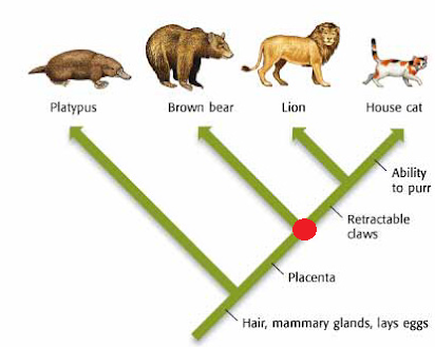
On the right is an example of a phylogenetic tree based on a few distinct biological features found in mammals. Here we see the most recent common ancestor of the brown bear, lion, and house cat, marked with a red dot. This ancestor represents the differentiation point between its descendants with and without retractable claws. From the common ancestor to the brown bear, no species developed retractable claws. Whereas from the common ancestor to the lion and house cat, retractable claws developed as a trait. This idea of definable traits can be applied to molecular and genetic traits as well. |
|
Phylogenetic Inference Methods
Maximum Likelihood:
|
|
With maximum likelihood testing, an initial tree is created using a faster, but more inaccurate method compared to other models. The data set and evolutionary model is used to make a maximized probability for the tree, which optimizes branch length and tree topology.
|
Neighbor Joining:
|
|
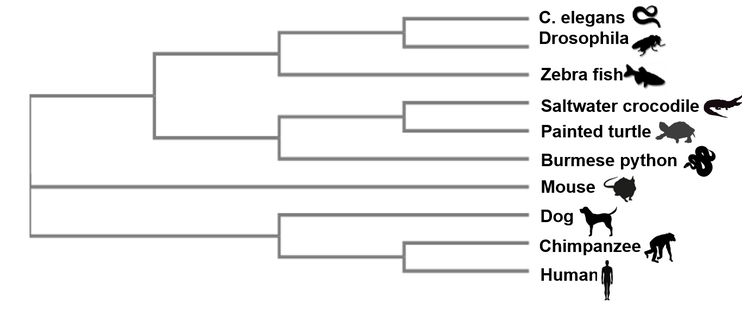
The neighbor joining (NJ) method is useful for analyzing species with highly varying lineages. This approach uses similarity scores to give generally fast and accurate results. However, neighbor joining only creates a single tree and does no consider other analysis methods.
|
Discussion |
|
The neighbor joining phylogeny method is a good model to show the ancientness and high conservation of the ABCA12 gene. Across terrestrial, aquatic, mammalian, and non-mammalian organisms, the ABCA12 gene is found in the genome.
|
|
References:
[1]. Reading a Phylogenetic Tree. Retreived from https://www.nature.com/scitable/topicpage/reading-a-phylogenetic-tree-the-meaning-of-41956/ |
Reference Images:
Mammal Phylogeny: Edited from https://tse3.mm.bing.net/th?id=OIP.LEBzieEu6FrsaD_RS6yxqwHaGR&pid=Api&P=0&w=300&h=300 Maximum Likelihood Phylogeny: Produced from http://www.phylogeny.fr/index.cgi Neighbor Joining Phylogeny: Produced from https://www.ebi.ac.uk/Tools/msa/clustalo/ |
This web page was produced as an assignment for Genetics 564, an undergraduate capstone course at UW-Madison
